
Übersichtsartikel von Leslie Magtanong und Scott J. Dixon
Eine neue Form von Zelltod
Der Zelltod ist ein wesentlicher Bestandteil der normalen Entwicklung und Aufrechterhaltung der Homöostase während des gesamten Lebens eines Organismus. Der Zelltod kann durch Caspase-vermittelte Apoptose oder durch eine wachsende Anzahl von nicht-apoptotischen Prozessen erfolgen. Diese nicht apoptotischen Prozesse umfassen Nekroptose, Pyroptose, Parthanatos und weitere. 1 Ferroptose ist eine der erst kürzlich beschriebenen Formen des nicht-apoptotischen Zelltods.2 Es ist ein oxidativer, eisenabhängiger Prozess, der durch das Anhäufen von lipid-reaktiven Sauerstoffspezies (ROS) auf ein toxisches Niveau entsteht.3,4 Allem Anschein nach erfordert die Ferroptose kein bestimmtes Effektorprotein (z.B. ein porenbildendes Protein). Dieser Prozess steht damit im Gegensatz zur Apoptose und anderen Formen des nicht-apoptotischen Zelltodes. Vielmehr konzentriert sich dieser tödliche Prozess auf die eisenabhängige, oxidative Zerstörung von Membranlipiden.
Wie der Name schon sagt, wird der ferroptotische Zelltod durch den Eisenbedarf definiert. Extrazelluläres Eisen wird durch das Protein Transferrin gebunden und durch den Transferrin-Rezeptor in die Zelle transportiert.5,6 Eisen wird im Lysosom aus dem Transferrin/Transferrin-Rezeptor-Komplex in das Zytoplasma freigesetzt.5,6 Innerhalb des Zytosols kann freies Eisen an der Fenton-Reaktion teilnehmen, wodurch Hydroxylradikale entstehen. Diese Radikale können dann mit mehrfach ungesättigten Acylketten in Membranphospholipiden reagieren. Hierdurch entstehen Lipidradikale, die leicht von Sauerstoff angegriffen werden, so dass sich hochreaktive Lipidperoxylradikale bilden. Diese Radikale können Protonen aus benachbarten Phospholipiden abstoßen, was zur Bildung weiterer Lipidperoxide und zur Ausbreitung von Membranschäden führt. Dieser Prozess der Autoxidation während der Ferroptose kann ausreichen, um eine offene Membranpermeabilisierung in einigen Zellen zu bewirken.7 In anderen hingegen ist u.a. auch die enzymatische Oxidation von mehrfach ungesättigten Fettsäuren (Poly Unsaturated Fatty Acids = PUFAs) durch eisenhaltige 12/15-Lipoxygenase-(LO/LOX)-Enzyme an dem Prozess beteiligt.8-10
Die Rolle von GPX4
In der Regel werden hochreaktive Lipidperoxide durch die Aktivität des essentiellen Enzyms Glutathionhydroperoxidase 4 (GPX4) zu nicht-reaktiven Lipidalkoholen reduziert. GPX4 ist ein Selenocystein-haltiges Enzym, das die reduzierte Form des Glutathions (GSH) für seine Aktivität benötigt.11
Cystein ist das geschwindigkeitsbestimmende Substrat bei der ATP-abhänigen GSH-Biosynthese, bei dem Cystein mit Glutamat und Glycin verbunden wird. Cystein kann aus Methionin synthetisiert werden (durch Transsulfurierung). Es kann aber auch als Cystin (Cys2, die oxidierte Form von Cystein) in die Zelle eingeführt werden. Das passiert durch einen natriumunabhängigen Antiporter namens System xc-. System xc- ist ein Heterodimer. Es besteht aus einer schweren Kette xCT (kodiert durch SLC7A11) und einer leichten Kette 4F2 (kodiert durch SLC3A2).12
Dieser Antiporter tauscht intrazelluläres Glutamat gegen extrazelluläres Cys2 im Verhältnis 1:1 aus. Im Cytosol angekommen wird das Cystin zu Cystein reduziert, welches in den Biosynthesepfad des Glutathions gelangt. Durch das Aufrechterhalten der intrazellulären Cystein-Konzentration ist die System xc--Aktivität ein entscheidender Faktor, um Ferroptose in vielen Zellen zu verhindern. Wenn der GSH-Spiegel unter einen bestimmten Schwellenwert fällt, ist GPX4 nicht mehr aktiv. Nun können sich die Lipid-ROS zu toxischen Konzentrationen anreichern, wodurch die Zelle einen ferroptotischen Zelltod stirbt.2,13
 Ferroptose kann durch Hemmung der xc--Glutathion-GPX4-Signalweges, an verschiedenen Stellen ausgelöst werden. Kleine Moleküle, die den Glutathionspiegel senken, werden als Ferroptose-induzierende Verbindungen (FINs) der Klasse I bezeichnet. Diejenigen, die Ferroptose durch direkte Hemmung von GPX4 ohne Glutathionabbau verursachen, werden als FINs der Klasse II bezeichnet.14 Erachin (FIN I) wurde ursprünglich durch die Expression des Onkogens HRASV12 als selektiv tödlich für künstlich veränderte Tumorzellen beschrieben.15,16 Erastin (und Analoga), der Tyrosinkinase-Hemmer Sorafenib (BAY 43-9006) und das Rheumamittel Sulfasalazin können das System xc- hemmen. Dadurch sinkt der GSH-Spiegel und Ferroptose wird ausgelöst.2,17 Klasse-II-FINs wie (1S,3R)-RSL3, ML-162 u.ä. Moleküle binden kovalent an GPX4 und inaktivieren es.8,14 Das Endoperoxid FINO2 fördert die Ferroptose, indem es die GPX4-Aktivität verringert und Eisen(III)-oxid oxidiert.18 Schließlich induziert das oximhaltige Molekül FIN56 Ferroptose, indem es den GPX4-Proteinspiegel reduziert und auch die Synthese des endogenen lipophilen Antioxidans-Metaboliten Coenzym Q10 (CoQ10) stört.19 Andere Moleküle, wie Statine, können auch indirekt Ferroptose induzieren, indem sie GPX4 destabilisieren und die CoQ10-Synthese hemmen.20 Obwohl Klasse-I-FINs als Induktoren der Ferroptose in Krebszellen in vivo vielversprechend sind, ist die Entwicklung von Analogen mit noch höherer Wirksamkeit in vivo äußerst wichtig.20,22,23 Bisher hat jedoch keines der bestehenden Klasse-II-FINs eine in vivo-Aktivität gezeigt. Die Entwicklung solcher Wirkstoffe wird notwendig sein, um das Potenzial für die gezielte Anwendung von GPX4 bei Krebs in vivo weiter zu untersuchen. Andere Mittel, wie eisenbindende Nanopartikel, können ein alternatives Mittel zur Induktion von Ferroptose in vivo sein.24
Ferroptose kann durch Hemmung der xc--Glutathion-GPX4-Signalweges, an verschiedenen Stellen ausgelöst werden. Kleine Moleküle, die den Glutathionspiegel senken, werden als Ferroptose-induzierende Verbindungen (FINs) der Klasse I bezeichnet. Diejenigen, die Ferroptose durch direkte Hemmung von GPX4 ohne Glutathionabbau verursachen, werden als FINs der Klasse II bezeichnet.14 Erachin (FIN I) wurde ursprünglich durch die Expression des Onkogens HRASV12 als selektiv tödlich für künstlich veränderte Tumorzellen beschrieben.15,16 Erastin (und Analoga), der Tyrosinkinase-Hemmer Sorafenib (BAY 43-9006) und das Rheumamittel Sulfasalazin können das System xc- hemmen. Dadurch sinkt der GSH-Spiegel und Ferroptose wird ausgelöst.2,17 Klasse-II-FINs wie (1S,3R)-RSL3, ML-162 u.ä. Moleküle binden kovalent an GPX4 und inaktivieren es.8,14 Das Endoperoxid FINO2 fördert die Ferroptose, indem es die GPX4-Aktivität verringert und Eisen(III)-oxid oxidiert.18 Schließlich induziert das oximhaltige Molekül FIN56 Ferroptose, indem es den GPX4-Proteinspiegel reduziert und auch die Synthese des endogenen lipophilen Antioxidans-Metaboliten Coenzym Q10 (CoQ10) stört.19 Andere Moleküle, wie Statine, können auch indirekt Ferroptose induzieren, indem sie GPX4 destabilisieren und die CoQ10-Synthese hemmen.20 Obwohl Klasse-I-FINs als Induktoren der Ferroptose in Krebszellen in vivo vielversprechend sind, ist die Entwicklung von Analogen mit noch höherer Wirksamkeit in vivo äußerst wichtig.20,22,23 Bisher hat jedoch keines der bestehenden Klasse-II-FINs eine in vivo-Aktivität gezeigt. Die Entwicklung solcher Wirkstoffe wird notwendig sein, um das Potenzial für die gezielte Anwendung von GPX4 bei Krebs in vivo weiter zu untersuchen. Andere Mittel, wie eisenbindende Nanopartikel, können ein alternatives Mittel zur Induktion von Ferroptose in vivo sein.24
Ferroptose-Inhibierung
Die Begrenzung der Lipid-ROS-Anreicherung verhindert Ferroptose, auch wenn GSH aufgebraucht oder GPX4 inaktiviert ist. Angesichts der zentralen Bedeutung von eisen- und membrangebundenen PFUAs können Mutationen als potente Inhibitoren der Ferroptose wirken. Die Mutationen sollen entweder zu einer geringeren Eisenaufnahme oder zum Stören des Einbaus von Lipiden in Zellmembranen führen. So begrenzt das genetische Silencing des Transferrin-Rezeptors die Eisenakkumulation und die Induktion von Ferroptose.25,26 Die Integration von Eisen in Eisen-Schwefel-Cluster dient auch als endogener Mechanismus zur Begrenzung der freien Eisenansammlung im Zytosol. Dies ist besonders wichtig, um die Ferroptose während der Hypoxie zu begrenzen.27 Die Aufnahme von PUFAs in Membranlipide erfordert zunächst die Aktivierung freier Fettsäuren. Sie werden durch die Acyl-CoA-Synthase Long chain member 4 (ACSL4) in PUFA-CoAs umgewandelt. Anschließend erfolgt die Aufnahme des PUFA-CoA in Lipide durch die Lysophosphatidylcholinacyltransferase 3 (LPCAT3).2,28-30 Die genetische Störung dieser Enzyme kann daher den Zelltod unter Ferroptose-induzierenden Bedingungen inhibieren. Dazu werden für die Durchführung dieses Prozesses erforderliche Schlüsselsubstrate (PUFA-haltige Lipide) entfernt. Naturstoffe, synthetische niedermolekulare Eisenchelatoren und lipophile Antioxidantien sind ebenfalls wirksame Ferroptose-Inhibitoren, die durch beide Klassen von FINs angeregt werden.4
Deferoxamin ist ein bakterieller Siderophor, der dreiwertiges Eisen chelatisiert. Es war eines der ersten bekannten Moleküle, das die Ferroptose hemmt.2,25 Lipophile Antioxidantien wie Ferrostatin-1 und Liproxstatin-1 können auch alle Ferroptosen in vitro und mit unterschiedlicher Wirksamkeit in vivo blockieren. 2,13,31,32 Ferrostatin-1 und Liproxstatin-1 sind beides Diarylamine. Mechanistisch gesehen ergibt sich die antioxidative Aktivität aus ihrer Fähigkeit, Acylketten tragende Peroxylradikale in Lipiddoppelschichten zu binden.33-35 Andere kleine Moleküle wie Vitamin E-Hydrochinon können Ferroptose verhindern, indem sie die Funktion von LO-Enzymen hemmen.36 Nicht zuletzt kann auch die Störung der Umwandlung von Glutamin in Glutamat durch Compound 968 Ferroptose in einigen Modellen teilweise inhibieren. Leider ist der biochemische Mechanismus, der Glutamin mit Eisen und/oder Lipid-ROS verbindet, nach wie vor unzureichend geklärt.26 Dennoch konnten diese Ferroptose-Inhibitoren in einem breiten Spektrum von ex vivo und in vivo Modellen des akuten und chronischen pathologischen Zelltods nachgewiesen werden.2,13,26,32 In der Zukunft der Ferroptose-Forschung bedarf es weiteren Tests mit komplexen Krankheitsmodellen oder humanklinischen Studien.
Finde Auslöser der Ferroptose
FSP1 Fluorescent Inhibitor Screening Assay Kit - NEU
Caymans FSP1 Fluorescent Inhibitor Screening Assay Kit bietet eine robuste und einfach zu verwendende Plattform zur Identifizierung neuer Inhibitoren von humanem FSP1, einem negativen Regulator des Ferroptose-Signalwegs.
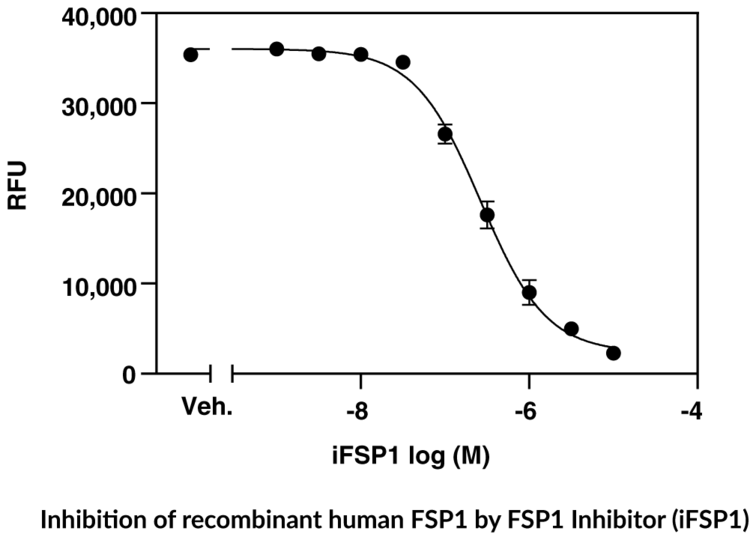
- Screene nach Inhibitoren von FSP1
- Enthält rekombinantes humanes FSP1 und den FSP1-Inhibitor iFSP1 als Positivkontrolle
- Testen Sie 45 Proben in zweifacher oder 29 Proben in dreifacher Ausführung
- Plattenbasierte fluorometrische Messung (Ex/Em = 540/590 nm)
GPX4 Inhibitor Screening Assay Kit - NEU
Caymans GPX4 Inhibitor Screening Assay Kit bietet eine robuste und einfach zu verwendende Plattform zur Identifizierung neuer Inhibitoren von humanem GPX4, einem negativen Regulator des Ferroptose-Signalwegs.
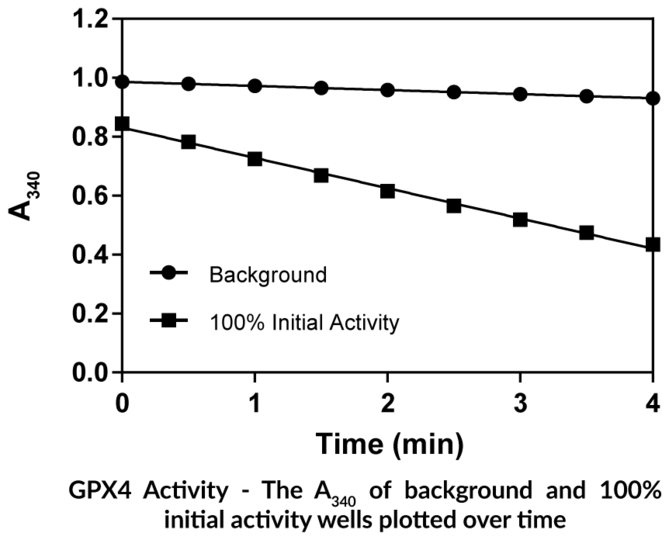
- Screene nach Inhibitoren von GPX4
- Enthält rekombinantes humanes GPX4
- Testen Sie 45 Proben in zweifacher oder 29 Proben in dreifacher Ausführung
- Plattenbasierte kolorimetrische Messung (340 nm)
Referenzen
- Galluzzi, L., Vitale, I., Aaronson, S.A., et al. Cell Death Differ. 25(3), 486-541 (2018).
- Dixon, S.J., Lemberg, K.M., Lamprecht, M.R., et al. Cell 149(5), 1060-1072 (2012).
- Cao, J.Y. and Dixon, S.J. Cell. Mol. Life Sci. 73(11-12), 2195-2209 (2016).
- Stockwell, B.R., Friedmann Angeli, J.P., Bayir, H., et al. Cell 171(2), 273-285 (2017).
- Andrews, N.C. and Schmidt, P.J. Annu. Rev. Physiol. 69(1), 69-85 (2007).
- Kawabata, H. Free Radic. Biol. Med. (2018).
- Shah, R., Shchepinov, M.S., and Pratt, D.A. ACS Cent. Sci. 4(3), 387-396 (2018).
- Yang, W.S., Kim, K.J., Gaschler, M.M., et al. Proc. Natl. Acad. Sci. U.S.A. 113(34), E4966-E4975 (2016).
- Shintoku, R., Takigawa, Y., Yamada, K., et al. Cancer Sci. 108(11), 2187-2194 (2017).
- Wenzel, S.E., Tyurina, Y.Y., Zhao, J., et al. Cell 171(3), 628-641 (2017).
- Ingold, I., Berndt, C., Schmitt, S., et al. Cell 172(3), 409-422 (2018).
- Lewerenz, J., Hewett, S.J., Huang, Y., et al. Antioxid. Redox Signal. 18(5), 522-555 (2013).
- Skouta, R., Dixon, S.J., Wang, J., et al. J. Am. Chem. Soc. 136(12), 4551-4556 (2014).
- Yang, W.S., SriRamaratnam, R., Welsch, M.E., et al. Cell 156(1-2), 317-331 (2014).
- Dolma, S., Lessnick, S.L., Hahn, W.C., et al. Cancer Cell 3(3), 285-296 (2003).
- Yagoda, N., von Rechenberg, M., Zaganjor, E., et al. Nature 447(7146), 864-868 (2007).
- Dixon, S.J., Patel, D.N., Welsch, M., et al. Elife 3, e02523 (2014).
- Gaschler, M.M., Andia, A.A., Liu, H., et al. Nat. Chem. Biol. 14(5), 507-515 (2018).
- Shimada, K., Skouta, R., Kaplan, A., et al. Nat. Chem. Biol. 12(7), 497–503 (2016).
- Viswanathan, V.S., Ryan, M.J., Dhruv, H.D., et al. Nature 547(7664), 453-457 (2017).
- Larraufie, M.-H., Yang, W.S., Jiang, E., et al. Bioorg. Med. Chem. Lett. 25(21), 4787-4792 (2015).
- Tsoi, J., Robert, L., Paraiso, K., et al. Cancer Cell 33(5), 890-904 (2018).
- Hangauer, M.J., Viswanathan, V.S., Ryan, M.J., et al. Nature 551(7679), 247-250 (2017).
- Kim, S.E., Zhang, L., Ma, K., et al. Nat. Nanotechnol. 11(11), 977-985 (2016).
- Yang, W.S. and Stockwell, B.R. Chem. Biol. 15(3), 234-245 (2008).
- Gao, M., Monian, P., Quadri, N., et al. Mol. Cell 59(2), 298-308 (2015).
- Alvarez, S.W., Sviderskiy, V.O., Terzi, E.M., et al. Nature 551(7682), 639-643 (2017).
- Dixon, S.J., Winter, G.E., Musavi, L.S., et al. ACS Chem. Biol. 10(7), 1604-1609 (2015).
- Kagan, V.E., Mao, G., Qu, F., et al. Nat. Chem. Biol. 13(1), 81-90 (2017).
- Doll, S., Proneth, B., Tyurina, Y.Y., et al. Nat. Chem. Biol. 13(1), 91-98 (2017).
- Friedmann Angeli, J.P., Schneider, M., Proneth, B., et al. Nat. Cell Biol. 16(12), 1180-1191 (2014).
- Linkermann, A., Skouta, R., Himmerkus, N., et al. Proc. Natl. Acad. Sci. U.S.A. 111(47), 16836-16841 (2014).
- Zilka, O., Shah, R., Li, B., et al. ACS Cent. Sci. 3(3), 232-243 (2017).
- Shah, R., Margison, K., and Pratt, D.A. ACS Chem. Biol. 12(10), 2538-2545 (2017).
- Poon, J.-F. and Pratt, D.A. Acc. Chem. Res. 51(9), 1996-2005 (2018).
- Hinman, A., Holst, C.R., Latham, J.C., et al. PLoS One 13(8), e0201369 (2018).


 Assay Kit")

-Sulfoximine")
")

-RSL3")



-HpETE-sn-glycero-3-PE")
-HETE-sn-glycero-3-PE")
")

